
The submission dossier is the packet of documents that are to be submitted to a health authority for registration of a product, or for other life-cycle maintenance activities, such as renewal of registration or CMC variations.
The requirements vary very widely from country to country, but in general a dossier contains administrative documents, (such as forms) legal documents, (contracts, authorizations, letters of representation, intelectual property documents, etc) technical CMC -Chemistry, Manufacturing and Control- documents, (quality specifications and testing standarts, list of ingredients, manufacturing description, validations, etc) clinical and/or preclinical documents, (clinical protocols, clinical trial results, patient informed consent documents, etc) labeling documents. (patient information leaflet, packaging artwork and labeling, physician’s information)
Some countries request the dossier to be structured with a specific format and layout, such as CTD or ASEAN dossier, whereas some other countries only request a list of documents without any specification on presentation. Some countries require or prefer electronic submission.
CTD: Common Technical Document
The CTD is a set of specifications for submission dossier for the registration of medicines. and designed to be used across Europe, Japan and the United States. It is an internationally agreed format developed by the European Medicines Agency, the Food and Drug Administration (U.S.) and the Ministry of Health, Labour and Welfare (Japan). It is maintained by the ICH. After the United States, the EU and Japan, the CTD has been adopted by several other countries including Canada, Switzerland, Australia, New Zealand, India, and China.
The agreement to assemble all the Quality, Safety and Efficacy information in a common format has revolutionised the regulatory review processes, led to harmonised electronic submission that, in turn, enabled implementation of good review practices. For industries, it has eliminated the need to reformat the information for submission to the different ICH regulatory authorities. (from ICH website)
This figure below is a schematic representation of the structure of the CTD.
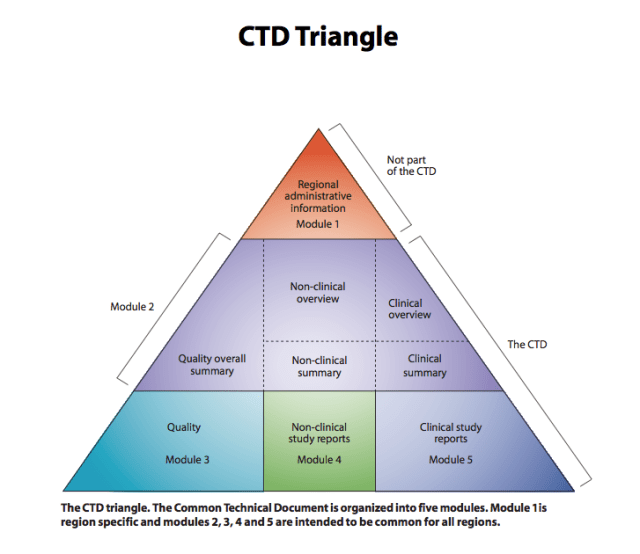
from http://www.ich.org
The eCTD is the electronic version of this dossier format.
CTD Format: module numbering and section heading.
1 Administrative and Product Information
Module 1 is not harmonized, its content will depend on the country or region in which the product is intended to be registered. It includes local forms and more administrative and legal documents and information.
2 Common Technical Document Summaries
2.1 Common Technical Document Table of Contents (Modules 2-5)
2.2 Introduction
2.3 Quality Overall Summary
2.4 Nonclinical Overview
2.5 Clinical Overview
2.6 Nonclinical Summary and Tabulated Summaries
2.7 Clinical Summary
3 Quality
3.1 Table of Contents of Module 3
3.2 Body of Data
3.2.S Drug Substance
3.2.S.1 General Information
3.2.S.1.1 Nomenclature
3.2.S.1.2 Structure
3.2.S.1.3 General Properties
3.2.S.2 Manufacture
3.2.S.2.1 Manufacturer(s)
3.2.S.2.2 Description of Manufacturing Process and Process Controls
3.2.S.2.3 Control of Materials
3.2.S.2.4 Controls of Critical Steps and Intermediates
3.2.S.2.5 Process Validation and/or Evaluation
3.2.S.2.6 Manufacturing Process Development
3.2.S.3 Characterisation
3.2.S.3.1 Elucidation of Structure and other Characteristics
3.2.S.3.2 Impurities
3.2.S.4 Control of Drug Substance
3.2.S.4.1 Specification
3.2.S.4.2 Analytical Procedures
3.2.S.4.3 Validation of Analytical Procedures
3.2.S.4.4 Batch Analyses
3.2.S.4.5 Justification of Specification
3.2.S.5 Reference Standards or Materials
3.2.S.6 Container Closure System
3.2.S.7 Stability
3.2.S.7.1 Stability Summary and Conclusions
3.2.S.7.2 Post-approval Stability Protocol and Stability Commitment
3.2.S.7.3 Stability Data
3.2.P Drug Product
3.2.P.1 Description and Composition of the Drug Product
3.2.P.2 Pharmaceutical Development
3.2.P.3 Manufacture
3.2.P.3.1 Manufacturer(s)
3.2.P.3.2 Batch Formula
3.2.P.3.3 Description of Manufacturing Process and Process Controls
3.2.P.3.4 Controls of Critical Steps and Intermediates
3.2.P.3.5 Process Validation and/or Evaluation
3.2.P.4 Control of Excipients
3.2.P.4.1 Specifications
3.2.P.4.2 Analytical Procedures
3.2.P.4.3 Validation of Analytical Procedures
3.2.P.4.4 Justification of Specifications
3.2.P.4.5 Excipients of Human or Animal Origin
3.2.P.4.6 Novel Excipients
3.2.P.5 Control of Drug Product
3.2.P.5.1 Specification(s)
3.2.P.5.2 Analytical Procedures
3.2.P.5.3 Validation of Analytical Procedures
3.2.P.5.4 Batch Analyses (For BGTD): Batch Analyses (For TPD):
3.2.P.5.5 Characterisation of Impurities
3.2.P.5.6 Justification of Specification(s)
3.2.P.6 Reference Standards or Materials
3.2.P.7 Container Closure System
3.2.P.8 Stability
3.2.P.8.1 Stability Summary and Conclusions
3.2.P.8.2 Post-approval Stability Protocol and Stability Commitment
3.2.P.8.3 Stability Data
3.2.A Appendices
3.2A.1 Facilities and Equipment
3.2.A.2 Adventitious Agents Safety Evaluation
3.2.A.3 Excipients
3.2.R Regional Information
3.2.R.1 Production Documentation
3.2.R.2 Medical Devices
3.2.R.3 Lot Release Documentation (for BGTD)
3.2.R.4 Yearly Biologic Product Reports (BGTD only)
3.3 Literature References
4 Nonclinical Study Reports
4.1 Table of Contents of Module 4
4.2 Study Reports
4.2.1 Pharmacology
4.2.1.1 Primary Pharmacodynamics
4.2.1.2 Secondary Pharmacodynamics
4.2.1.3 Safety Pharmacology
4.2.1.4 Pharmacodynamic Drug Interactions
4.2.2 Pharmacokinetics
4.2.2.1 Analytical Methods and Validation Reports (if separate reports are available)
4.2.2.2 Absorption
4.2.2.3 Distribution
4.2.2.4 Metabolism
4.2.2.5 Excretion
4.2.2.6 Pharmacokinetic Drug Interactions (nonclinical)
4.2.2.7 Other Pharmacokinetic Studies
4.2.3 Toxicology
4.2.3.1 Single-Dose Toxicity (in order by species, by route)
4.2.3.2 Repeat-Dose Toxicity
4.2.3.3 Genotoxicity
4.2.3.3.1 In vitro
4.2.3.3.2 In vivo (supportive toxicokinetics evaluations)
4.2.3.4 Carcinogenicity (including toxicokinetics)
4.2.3.4.1 Long-term studies (not included in repeat-dose toxicity or pharmacokinetics)
4.2.3.4.2 Short- or medium-term studies (not included under repeat-dose toxicity or pharmacokinetics
4.2.3.4.3 Other studies
4.2.3.5 Reproductive and Developmental Toxicity
4.2.3.5.1 Fertility and early embryonic development
4.2.3.5.2 Embryo-fetal development
4.2.3.5.3 Prenatal and postnatal development, including maternal function
4.2.3.5.4 Studies in which the offspring (juvenile animals) are dosed and/or further evaluated
4.2.3.6 Local Tolerance
4.2.3.7 Other Toxicity Studies (if available)
4.2.3.7.1 Antigenicity
4.2.3.7.2 Immunotoxicity
4.2.3.7.3 Mechanistic studies (if not included elsewhere)
4.2.3.7.4 Dependence
4.2.3.7.5 Metabolites
4.2.3.7.6 Impurities
4.2.3.7.7 Other
4.3 Literature References
5 Clinical Study Reports
5.1 Table of Contents of Module 5
5.2 Tabular Listing of Clinical Studies
5.3 Clinical Study Reports
5.3.1 Reports of Biopharmaceutic Studies
5.3.1.1 Bioavailability (BA) Study Reports
5.3.1.2 Comparative Bioavailability (BA) and Bioequivalence (BE) Study Reports
5.3.1.3 In vitro-In vivo Correlation Study Reports
5.3.1.4 Reports of Bioanalytical and Analytical Methods for Human Studies
5.3.2 Reports of Studies Pertinent to Pharmacokinetics using Human Biomaterials
5.3.2.1 Plasma Protein Binding Study Reports
5.3.2.2 Reports of Hepatic Metabolism and Drug Interaction Studies
5.3.2.3 Reports of Studies Using Other Human Biomaterials
5.3.3 Reports of Human Pharmacokinetic (PK) Studies
5.3.3.1 Healthy Subject PK and Initial Tolerability Study Reports
5.3.3.2 Patient PK and Initial Tolerability Study Reports
5.3.3.3 Intrinsic Factor PK Study Reports
5.3.3.4 Extrinsic Factor PK Study Reports
5.3.3.5 Population PK Study Reports
5.3.4 Reports of Human Pharmacodynamic Studies
5.3.4.1 Healthy Subject PD and PK/PD Study Reports
5.3.4.2 Patient PD and PK/PD Study Reports
5.3.5 Reports of Efficacy and Safety Studies
5.3.5.1 Study Reports of Controlled Clinical Studies Pertinent to the Claimed Indication
5.3.5.2 Study Reports of Uncontrolled Clinical Studies References
5.3.5.3 Reports of Analyses of Data from more than one study, including any formal integrated analyses, meta-analyses, and bridging analyses
5.3.5.4 Other Clinical Study Reports
5.3.6 Reports of Postmarketing Experience
5.3.7 Case Report Forms and Individual Patient Listings (when submitted)
5.4 Literature References





Whatabout maintenance activites
Do type 1 and 2 variations apply in latin america?
Hello, Emma!
Thanks for reading my blog, for your interest and for your great question.
About maintenance activities:
1- Renewal of marketing authorization: generally, certificates have to be renewed every 5 years. There is great variability among countries on what a renewal requires, in some cases, such as Argentina, with the fees and some administrative information and documents is enough, in some other cases, such as Peru, all the registration dossier has to be submitted anew.
2- Neither there is regional harmonization on variations. Variations are not classified as 1 and 2. Variations apply when the modification affects any of the submitted technical or administrative documents. Each country has its own system, for example in Mexico variations are classified in technical and administrative, in Argentina in different categories that you can find here: https://latampharmara.com/argentina/variations/ and in some cases variations will signify new registrations, as is the case of Marketing Authorization Holder transfer in Brazil.
Hello, mari
for which category of product there is mandatory requirement of BA/BE studies in costarica and guatemal
vl u give me the comparision between ICH CTD Vs Argentina CTD
Hi Lokesh!
Thanks for your question. Argentina doesn’t require CTD. You can find the documents required for Argentina in this section: https://latampharmara.com/argentina/drug-registration-in-argentina/ You can extract the documents from the CTD, format (or rather, un-format them), translate them to Spanish and submit.
Have a nice weekend!
Maria
Hi Maria,
Please let me know the requirement of administrative documents and samples for the registration of the product in Central and Latin american countries.
Regards,
Ajay
CTD REQUIRE IN COSTA RICA? PLEASE PROVIDE ME GUILENRS FOR PRODUCT REGISTRATION IN COSTA RICA.
REGARDS,
PRAGNESH PATEL
Dear Pragnesh,
CTD is not required in Costa Rica, they rather ask for a list of specific documents, but don’t specify a format. I’m working right now in a section for Central America countries, please come back soon for updates.
Thanks and have a nice day!
Maria
Hi Maria,
Could u please tell me the procedure required to dispatch the CTD dossier for Latin America. Is it the same procedure as EU or its different.
I aslo want to know the difference between MHRA & eCTD submission (In naming convention of folder as well as document)
Dear Jagadhish,
Thank you for your interest and your question. The CTD is not required in Latin America. Each country has a specific list of technical documents they require, some of them in a specific format, some of them not, but in any case you will have to de-format the documents and re-customize them for each particular country. Also, you will need to translate the dossier to Portuguese for Brazil and to Spanish for the rest of the LatAm countries.
On your second question, unfortunately I’m not an expert in European dossier format 😦
Have a great weekend!
Maria
Dear Maria,
Would you please inform me about the dossier format for Dominican Republic??
Thanks for the information!
How Brazilian CDT requirements differ from ICH-CTD requirements?
will u give me info about the difference in USA and BRAZIL dossier
Hi Maria,
Greetings.
As i could see on this blog, that Panama registration requirements are under construction.
What I could gather for Panama filing-
1. CMC docs as per CTD
2. Stability- 3 batches, zone IV (30/65 Long term & 40/75 accelerated) stability studies
Please confirm these & also let me know:
1.What are BE requirements (if any specific), can it be done with other country innovator or from Panama only?
2. What are the requirements for process validation?
Thanks for your time.
Deepa
Hi Maria,
Is it possible for you to share the country specific guidelines for the following countries :
1.Costa rica
2.El Salvador
3.Panama
4.Guatemala
5.Ecuador
6.Honduras
I was trying to download from websites of ministry of health BUT everything is in spanish..would be great if you can help??
Thanks and Regards,
Rubiya
Hi, Is In LATAM countries dossier is mandatory in their country launguage or they can accpet in English?
Dear Maria
Can you provide me clinical and
nonclinical data for any product. (please help me. for clinical and non clinical)
with Regards
Manish bhatt